1. ISO 10993-1: 2018: Biological evaluation of medical devices as part of a risk management process
The ISO 10993-1: 2018 standard defines biocompatibility as the “ability of a medical device or material to perform with an appropriate host response in a specific application”.
Such a process should generally begin with assessment of the device, including
- material components,
- manufacturing processes,
- clinical use of the device including the intended anatomical location, and
- frequency and duration of exposure.
Considering this information, the potential risks from a biocompatibility perspective should be identified.
Such risks might include
- chemical toxicity
- unacceptable biological response to physical characteristics of the device, and
- aspects of manufacturing and processing that could alter the physicochemical characteristics of the device
Once the risks have been identified, the sponsor should assess what information is already available regarding those risks and identify the knowledge gaps that remain. Considering the potential biological impact, a plan should be developed to address the knowledge gaps either by biocompatibility testing or other evaluations that appropriately address the risks.
The interpretation of the overall biocompatibility evaluation should be considered in the appropriate benefit-risk context. *https://www.fda.gov/media/85865/download*
If the product has a direct or indirect contact with the patient, it is crucial to identify all possible information concerning the material in question (physico chemical properties, manufacturing processes).
If it is the same as in the marketed device, we need to do a toxicological risk assessment (Annex B).
If it is not the same and there is no toxicological data concerning this new material, then there is a need to perform more evaluation on the device based on the chemical nature of the components and the duration of contact, we select biological endpoints (Annex A) and more tests in order to perform the toxicological risk assessment.
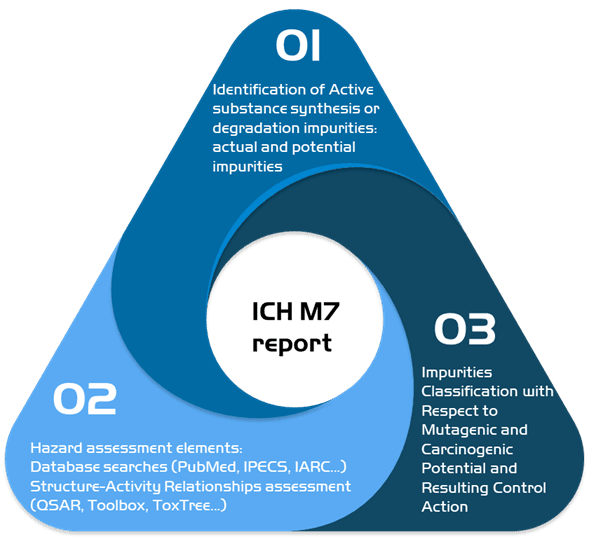
2. ICH M7: Development of the characterization of mutagenic impurities
In case you suspect the presence of mutagenic impurities, we can do an ICH M7 impurities risk assessment.
In our report, we identify all the active substances in the different stages of manufacturing in order to determine current and potential impurities, we then proceed to do a toxicological assessment of the elements found using our wide network of databases and using toxicological programs in order to classify the impurities found and recommend the proper control actions needed.
3. MEDDEV 2.7/1 revision 4 Guidelines on Medical Devices, Clinical Evaluation: A guide for manufacturers and notified bodies under directives 93/42/EEC and 90/385/EEC.
Common approach to clinical evaluation for medical devices
Attention: does not concern in vitro diagnostic devices.
The clinical evaluation is based on a comprehensive analysis of available pre- and post-market clinical data relevant to the intended purpose of the device in question, including clinical performance data and clinical safety data. There are discrete stages in performing an MD clinical evaluation: **
Stage 0: Define the scope, plan the clinical evaluation (also referred to as scoping and the clinical evaluation plan).
Stage 1: Identify pertinent data
Stage 2: Appraise each individual data set, in terms of its scientific validity, relevance and weighting
Stage 3: Analyse the data, whereby conclusions are reached about
- compliance with Essential Requirements (including ER1, ER3, ER6) on performance and safety of the device, including its benefit/risk profile,
- the contents of information materials supplied by the manufacturer (including the label, IFU of the device, available promotional materials, including accompanying documents possibly foreseen by the manufacturer),
- residual risks and uncertainties or unanswered questions (including on rare complications, long term performance, safety under wide-spread use), whether these are acceptable for CE-marking, and whether they are required to be addressed during "PMS" (Post-Market Surveillance)
Stage 4: Finalise the clinical evaluation report
The clinical evaluation is actively updated: When the manufacturer receives new information from Post Marketing Surveillance that has the potential to change the current evaluation;
if no such information is received, then at least annually if the device carries significant risks or is not yet well established; or every 2 to 5 years if the device is not expected to carry significant risks and is well established, a justification should be provided.
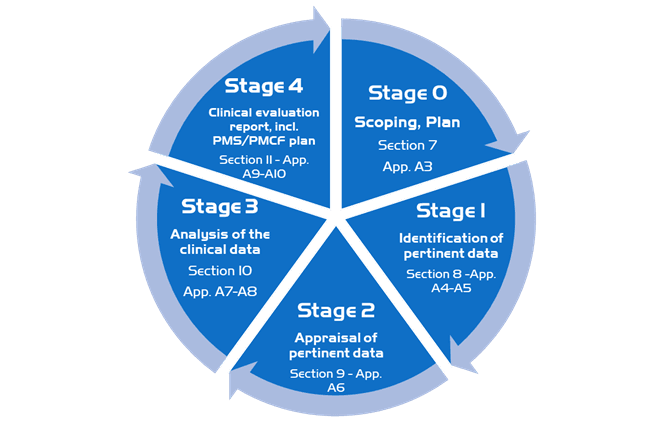
In our methodology, we follow the stages detailed in the guideline, we first put in place a scoping plan then identify and appraise important and relevant data and then analyze the clinical data in order to finalize the clinical evaluation report.


You can check Tox by Design methodology for Medical Devices Risk Assessments, signed by an European Registered Toxicologist expert here.

In the event you are performing this exercise for innovative compounds GMP scale up manufacturing, please be noted Tox by Design is duly accredited for the French Research Tax Credit CIR.
Feel free to contact us using this email link to receive a quotation for Tox Medical Devices procedure used on your manufacturing line.